ADEPT2 Trace Archives

241,796 reads submitted to Trace Archive
- 4 files per read
- Almost a million files
- Original chromat files with peak, qual, and fasta
- 18 GB of compressed data
- Submitted as 11 data sets
- Total of 7535 amplicons
Procedure overview:
- Pull data out of TreeGenes Database for reads marked as successful.
- Group reads according to amplicon.
- Determine amplification primer for opposite strand. If not present, return to database and get amplification primer based on additional info.
- Determine and confirm location of the 241,796 files on the loblolly server.
- Group amplicons so each submission will be ~1.5 GB when compressed.
- Create each data set with proper file structure.
- Generate fasta, qual and peak files in proper directories.
- Generate TRACEINFO.txt and MC5 information.
Each accepted read assigned a Trace Archive accession number.
- Treegenes database was modified to incorporate Trace Archive accession numbers for each successful read submitted.
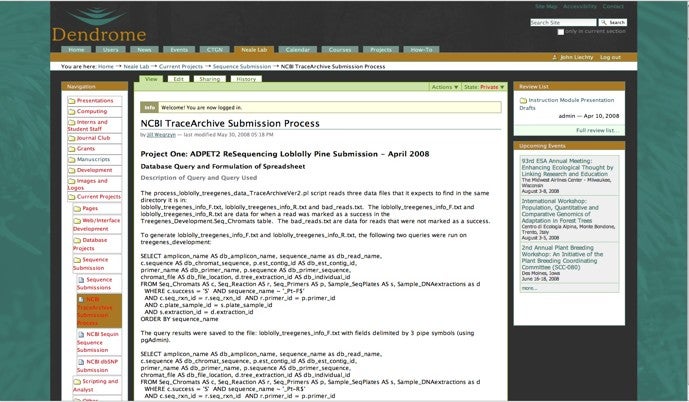
Supporting code and database queries have been documented on the Dendrome Plone content management content system.
- NCBI tags, SQL, scripts and procedures.
GenBank Submissions
6,178 PopSets submitted via NCBI\’s Sequin passed internal and external standards for acceptance
These sequence sets are accessible through the following accessions:
 dbSNP Submission
dbSNP Submission