ADEPT2 Phenotyping
Gene Expression Phenotyping and Association Genetics
 Nearly 1500 rooted cuttings were received from. North Carolina State University in spring 2006. That fall, they were harvested and roots, stems and needles were stored for RNA isolations. RNA was isolated from stems of two ramets per clone.
Nearly 1500 rooted cuttings were received from. North Carolina State University in spring 2006. That fall, they were harvested and roots, stems and needles were stored for RNA isolations. RNA was isolated from stems of two ramets per clone.Over 200 genes with potential roles in wood development or responses to biotic and abiotic stresses were chosen based on previous research in our laboratories and literature reviews. Real-time qPCR has been used to analyze 182 genes to date. The last 23 genes are currently being analyzed. We have discovered high levels of variation in gene expression between genotypes. The differences between high-expressing and low-expressing genotypes usually ranges between three and seven (rarely to 14) PCR cycles (usually 8 to 128 fold).
In many cases we observe strong correlations between the expression levels of various genes. In some cases, this suggests coordinate regulation. The DDCT values for phenylalanine ammonia lyase, a gene involved in lignin biosynthesis, and a myb transcription factor are compared below.
| Gene | Fold diff | Gene | Fold diff |
| CAD | 64 | Korrigan | 64 |
| AGP 1-6 | 16-64 | 4CL-1 | 64 |
| PAL-1 | 32 | Terp Syns | 32-128 |
| Suc Syn | 8 | C3H | 128 |
| Expansin 1 | 32 | PR-10 | 512 |
| MYB-1 | 32 | MYB- 4 | 32 |
| CeSAs 1-10 | 32-64 | ARF-11 | 32 |
| LEA-35 | 16,384 | Cyp B | 64 |
| Kobito | 32 | Tubulins | 8-16 |
| Cobra | 64 | CCR | 32 |
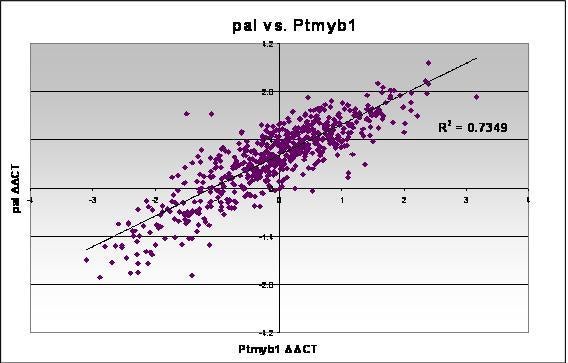
Figure 1. A loblolly pine myb promoter contains a SNP in a putative myb-binding site. CTAACCA – putative myb binding site TTAACCA – not predicted to be a myb binding site.
Such correlations caused us to clone promoters in search of SNPs in putative binding sites of trans-acting elements. Promoters from 13 transcription factors and three other genes induced by drought stress were cloned using a Genome Walker kit and sequenced. SNP discovery in the promoters is planned for this year in collaboration with UC-Davis. Four of the promoters were sequenced in a small number of individuals and an interesting SNP was found in the promoter of a myb gene induced by drought stress. One allele results in a putative binding site for another MYB with a known role in response to drought stress. The other allele does not have a putative binding site. In a preliminary experiment, four individuals from The Lost Pines, a drought-resistant population, contain one copy of the binding site while four individuals from Louisiana do not. This SNP was included on the Illumina chip.
Experiments in Progress
In addition to completing the remaining gene expression analyses and the promoter SNP genotyping, association analyses are just starting. One of the graduate students on the project, Sreenath Palle, went to UC-Davis in April 2008 to be trained in the use of the program Tassel?

With the exception of some LEA genes, most of the genes with potential roles in drought stress did not show much variation between clones. The plants had not been drought stressed before harvesting. The disease genes did not produce good quality data with differences between ramets, possibly due to a fungal infection in the greenhouse. Therefore, a previously unplanned, small experiment was initiated. 24 clones were selected for inclusion based on pitch canker data from the University of Florida and water use efficiency data from North Carolina State. The plants are currently ready to start a factorial experiment involving drought stress and inoculation with pitch canker. Three ramets per clone exposed to each of the four treatments (drought ?no disease; no drought ? no disease; drought ?pitch canker inoculation; no drought ?pitch canker inoculation) will have physiological traits measured and RNA isolated from the stems. qPCR will be performed to determine the expression of approximately 100 genes in the different clones and in response to the treatments.
Training Two Ph.D. students are being trained in the laboratory as part of this project. Both have completed all their coursework and have passed their preliminary examinations. Both are involved in the Graduate Student Teaching Academy to help prepare them to be the educators of the future. Sreenath Palle will graduate in August 2009 and Candace Seeve in August 2010. One visiting scientist from Nicaragua has been in the laboratory this semester and has received training in real-time qPCR, sequencing, and genotyping. Seven high school and middle school teachers from Texas were recruited and sent to the genomics workshop help at Clemson University.
- Metabolite Phenotyping and Association Genetics
- Wood Quality Phenotyping and Association Genetics
TASKS: Measure the wood mechanical, chemical and anatomical properties in a clonally propagated population. This structured CCLONES population contains 31 parents mated in a circular dialell. Approximately 15 progeny per cross were used to create hedges from which rooted cuttings were taken and planted in 2003. In all, 999 genotypes from 62 families are represented in the population that is being sampled. Four clonal replications per genotype (2 replications from each of 2 sites) have been cored (total of 3888 cores) and the wood properties are being measured from rings 3 and/or 4, depending on the property. Two significant challenges/set backs for the wood property phenotyping needed to be overcome. First, the closure of the wood x-ray micro ct instrument at Oakridge National Laboratory was over come with new funding to G. Peter from DOE which was used together with funds from this award to purchase an x-ray micro ct that is now up and running at UF. Second, the dramatic increase in cost by Silviscan for cellulose microfibril angle (MFA) measurement was overcome by moving to acoustic based approaches for measuring velocity wood stiffness as a surrogate for MFA, but very highly correlated trait.
MECHANICAL PROPERTIES
- For the 3888 trees in the study, in-tree velocity stiffness has been measured. As expected, wood stiffness shows a continuous distribution (Figure 1), indicating that it is inherited as a complex trait. The clonal repeatability, a measure of broad sense heritability for clones, for in-tree wood stiffness is 0.42, similar to other published results with slash and radiata pine families (Kumar et al., 2002; Kumar et al., 2006; Li et al., 2007), and has one of highest levels of genetic control of all wood properties. 65% of the genetic variation is additive demonstrating that breeding for improved juvenile wood stiffness will be quite successful. For a small sample of 60 trees, cellulose microfibril angle of the two outermost rings and in-tree velocity stiffness showed positive, genetic correlation (0.84).
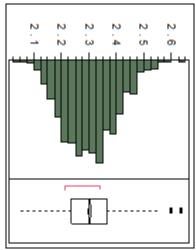
Figure 2. Distribution of LS means for In-tree velocity stiffness from 999 genotypes from two sites.
- In the lab, the velocity stiffness of the earlywood and latewood of rings 3 and 4 is being measured with a newly purchased and installed ultrasonic tester. Lab measurements of velocity from 5mm wood cores are being measured to confirm the rankings from the in-tree results and to relate stiffness to cellulose microfibril angle (MFA). Velocity measurements from the new ultrasonic tester and MFA measured with x-ray diffraction are highly, linearly correlated (r2=0.88) for both earlywood and latewood, demonstrating that velocity stiffness can be used to precisely estimate MFA. Velocity stiffness is a much more cost effective and higher throughput method compared with x-ray diffraction.
CHEMICAL PROPERTIES
- For all 3888 wood cores, wood chemical composition of rings 3 plus 4 has been measured by pyrolysis molecular beam mass spectrometry at the National Renewable Energy Lab, a method used previously for mapping QTL for these traits in loblolly pine (Sewell et al., 2002). Lignin, C5 and C6 sugar contents were inherited quantitatively with across site clonal repeatabilities of C5 ?0.11+0.03, C6- 0.14+0.03, and lignin ?0.13+0.02. Overall these show that wood chemical contents are under low genetic control in this population. The heritabilities found for CCLONES are slightly lower than the only published study for the genetic control of wood chemical content in pine; however the published study sampled substantially fewer trees and hence had large standard errors of the estimates (Sykes et al., 2006). C5 and C6 contents are strongly negatively genetically correlated with lignin content, -0.87 and -0.83, respectively. Interestingly, lignin content was positively correlated genetically with height and stem diameter growth, 0.67 and 0.57, respectively. Lignin content showed substantial genetic by environmental (GxE) interaction, while C6 showed little indication of GxE.

Figure 3. Cross sectional plane of wood core scanned with x-ray micro ct at 10 uM
ANATOMICAL PROPERTIES
-
- Wood density, tracheid cell dimensions, and lumen diameters in rings 3 and 4 are being measured using x-ray micro ct. An x-ray micro ct was purchased and recently installed. X-ray micro ct allows for low and high resolution scans and 3-dimensional tomographic reconstruction of the density profiles of wood. Wood density profiles will be measured at 74 uM resolution. From these profiles the average density and the percentage of both earlywood and latewood will be quantified for rings 3 and 4. Tracheid cell dimensions and lumen diameters will be quantified from 10 uM scans of ring 4.
Summary of wood property phenotyping in CCLONES.
| Wood Property | Status |
| Density | rings 3-4 in progress, expected 2/09 |
| % Latewood | rings 3-4 in progress, expected 2/09 |
| Tracheid dimensions | ring 4, expected 6/09 |
| Cellulose microfibril angle/velocity stiffness | rings 3-4 in progress, expected 1/09 |
| In-tree velocity stiffness | rings 3-4 ?complete |
| Lignin content | rings 3+4 ?complete |
| C6 content | rings 3+4 ?complete |
| C5 content | rings 3+4 ?complete |
- Disease Phenotyping and Association Genetics
TASKS: Measure the disease resistance of the unstructured NSF association population. This population contains 498 randomly selected trees from across the range of loblolly pine. Clonally propagated ramets were inoculated with spores from Fusarium circinatum and Cronartium quercuum f.sp. fusiforme and scored for lesion length and gall characteristics, respectively, as measures of resistance.
DISEASE PHENOTYPING FOR PITCH CANKER RESISTANCE
A population of 498 clonally propagated, unrelated genotypes (NSF association population) was screened for resistance to pitch canker under greenhouse conditions. Lesion length measurements were taken at 4, 8 and 12 weeks after inoculation. Figure below shows that resistance is a complex trait, which validates previous screens (Kayihan et al. 2005) implying that resistance is inherited quantitatively.
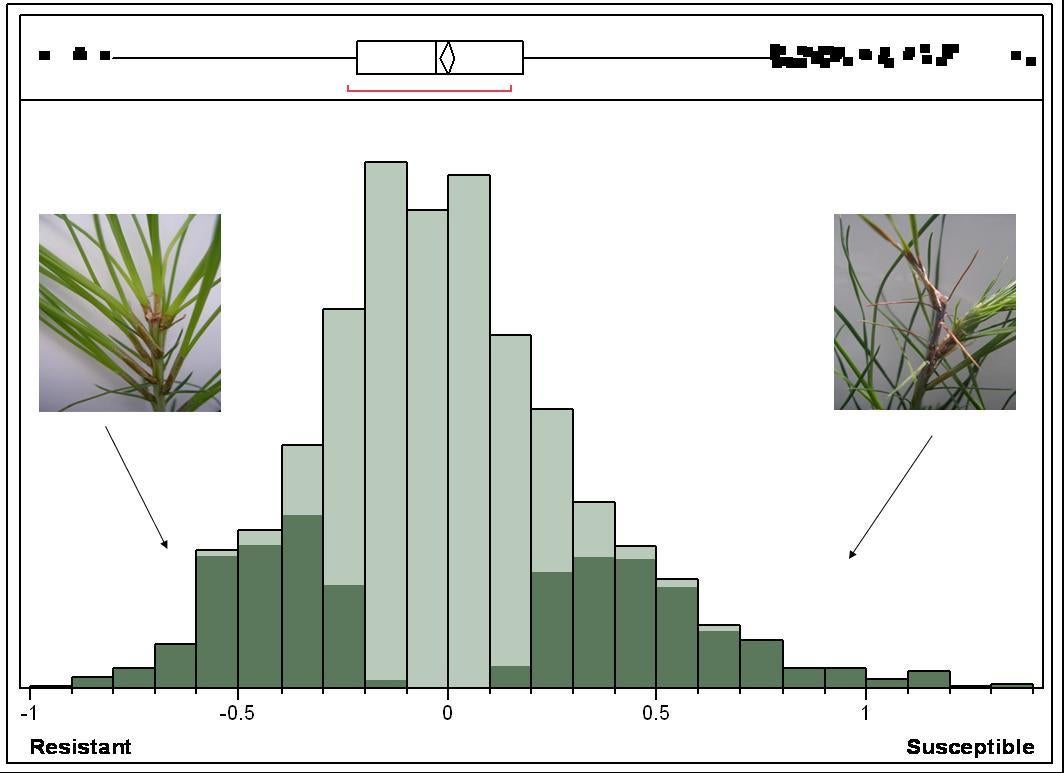
Figure 4. Distribution of BLUP clonal estimates for pitch canker lesion length (log-transformed), highlighting the 50 most resistant and susceptible clones with >0.3 cuttings. Inserts show examples of resistant and susceptible genotypes.
Genotypes with extreme resistance (45) and susceptibility (47) to pitch canker were selected based on their ranks from best linear unbiased predictions (BLUP) of clonal performance. Clonally propagated ramets were inoculated and used to confirm their rank positions in the primary screen.
Clonal values and variation for lesion length increased as disease progressed in all measurements, particularly in the susceptible tails. Significant differences between tails were observed (p<0.0001).
| 1st Inoculation (Association Population) |
2nd Inoculation (Tails) | ||||||||
| Resistant | Susceptible | ||||||||
| Time post-inoculation (weeks) | |||||||||
| 4 | 8 | 12 | 4 | 8 | 12 | 4 | 8 | 12 | |
| Mean(mm) | 5.75 | 7.93 | 9.50 | 2.86 | 3.81 | 4.57 | 7.05 | 12.15 | 14.01 |
| St. Dev. | 2.63 | 5.01 | 7.56 | 1.10 | 1.31 | 3.00 | 3.84 | 7.72 | 9.57 |
| Median | 5.37 | 6.67 | 7.27 | 2.69 | 3.38 | 3.87 | 6.31 | 10.05 | 10.75 |
| N | 498 | 45 | 47 | ||||||
Clonal repeatability, a measure of broad-sense heritability, was estimated for both the overall population as well as for the extreme genotypes (tails). These results compared well with previous genetic data obtained from the structured CCLONES population with known pedigree (Kayihan et al., 2005).
Clonal repeatability ranged between 0.21 and 0.28 in the first inoculation and between 0.35 and 0.39 in the second inoculation. Repeatabilities increased throughout measurements in both inoculation experiments. These results are comparable to the broad-sense heritability estimates reported by Kayihan et al. (2005) in a population with known pedigree (H2 = 0.37 ?0.43).
| Variances | ||||
| Lesion loglength | Clone | Ramet | Residual | Repeatability |
| First Inoculation | ||||
| 4 weeks | 0.11 | 0.098 | 0.32 | 0.21 |
| 8 weeks | 0.16 | 0.077 | 0.41 | 0.25 |
| 12 weeks | 0.21 | 0.069 | 0.47 | 0.28 |
| Second Inoculation | ||||
| 4 weeks | 0.29 | — | 0.54 | 0.35 |
| 8 weeks | 0.34 | — | 0.53 | 0.39 |
| 12 weeks | 0.35 | — | 0.56 | 0.38 |
DISEASE PHENOTYPING FOR FUSIFORM RUST RESISTANCE
A total of 497 genotypes were inoculated with three fungal cultures (SC 20-21, NC 2-36 or the P2 hybrid culture of the previous two) at the Resistance Screening Center in Asheville, NC in June, 2008. Four replicates were used for each inoculation, totaling 5,760 cuttings. Most genotypes (386) had enough cuttings (12) for inoculations with all 3 fungal cultures and 4 replicates. Clones that had between 3 and 11 cuttings were randomly assigned, such that at least one cutting of each clone was inoculated with each one of the 3 fungal cultures. The plants were transported to the University of Florida greenhouse facilities and were set in a random incomplete block design with four replicates. They are currently being scored for presence of galls. A collection of pycnial drops from the galls is also being established to obtain genotypic data of the fungus.
Summary of fungal disease phenotyping in NSF Association Population.
| Disease | Status |
| Pitch Canker | Completed |
| Fusiform rust | Expected 12/08 |
IDENTIFICATION OF POTENTIALLY SIGNIFICANT ASSOCIATIONS IN NSF ASSOCIATION POPULATION WITH PITCH CANKER RESISTANCE
A total of 3,938 SNP markers for 404 of the 498 clones in the NCSU association population were analyzed using the Bayesian method. Potentially significant associations with pitch canker resistance have been identified for 24 SNP markers in 24 genes. The EST sequences from the putative significant SNPs were obtained from the UGA and UMN databases, as well as from the Dendrome Plone.
A BLASTx search was performed against the entire NCBI protein database and the best hits were obtained. In cases where the best hit sequence from the database was an unknown protein, the next available sequence was considered until a best hit with known function was identified. This provided an idea of a possible protein function that may be influenced by SNP polymorphisms. The potential effects of SNP polymorphisms (changes in amino acids, premature termination codons, etc) are currently being assessed.
- Drought-Tolerance Phenotyping and Association Genetics
- Development of Association Genetic Methodologies
TASKS: Develop robust methods for detecting significant associations between SNP genotypes and phenotypes. The models must be capable of controlling for both population structures at a coarse level, such as those that arise from geographic location, and for fine population structures which arises from controlled mating designs. Construct a pipeline for handling data and running association analyses.
MODEL DEVELOPMENT AND VALIDATION WITH SIMULATIONS
A new Bayesian association framework has been developed and validated. The Bayesian framework accommodates analyses of unstructured and structured populations by implementing population structure and relationship matrices. Distinct advantages of this framework are that 1) simultaneous analysis of the set of SNPs is less biased and more likely to identify multiple causal SNPs, 2) informative priors can be assigned with the relationships are known. In addition, a Gibbs sampler has been implemented that imputes missing SNP data from all available relationship and phenotypic data, permitting the retention of more SNPs compared with mixed linear model methods. Sparse matrix technology together with improved programming methods has permitted the development of an R-package that is straightforward to use. The approach and program have been validated using simulated data. Using data simulated based on the CCLONES design; SNPs with small phenotypic effects were readily identified, even with 10% missing SNP data.
COMPARISON OF QTdT AND BAYESIAN MODEL WITH CCLONES SNP GENOTYPIC AND D13C PHENOTYPIC DATA
From previously funded ADEPT1research, 46 SNPs in candidate drought tolerance and disease resistance genes were genotyped in the CCLONES population and d13C carbon isotope discrimination data were collected from two sites (Gonzales-Martinez, et al., 2008). Using the quantitative transmission disequilibrium test (QTdT), 6 potential SNPs were identified as candidate genes controlling carbon isotope discrimination. With the Bayesian method with informative priors 4 of the 6 SNPs were identified.